JPM 2026 Day #3 Highlights –
Executive Highlights
- The third day of the 44th Annual JP Morgan Healthcare Conference continued strong at the Westin St. Francis, with topics ranging from obesity treatments to cell-based therapies for T1D, precision treatment for insulin-deficient T2D, and the ever-expanding role of digital health. Discussions on innovations, competitive market, and regulatory pathways built on insights from JPM Day #1 and Day #2.
- Curious what lies ahead tomorrow and want a reminder of all we’ve learned this week? See our preview and Resource Hub. We will be eager to hear what SAB Bio says - they are developing SAB-142 (human anti-thymocyte immunoglobulin) as a disease-modifying therapy for T1D. We’ll also hear from Lexicon, which is working on SGLT-1/2 inhibitor Zynquista and candidates for diabetic peripheral neuropathy and obesity, as well as Ypsomed, which recently launched new injection platforms YpsoFlow, YpsoDot, and YpsoLoop for drug delivery.
- In therapy, the obesity market continued to occupy a central role in presentations.
- Zealand CEO Adam Steensberg shared multiple insights about the obesity market and gave valuable therapy and updates on its pipeline, including survodutide (dual glucagon/GLP-1 RA) and petrelintide (long-acting amylin analog). He highlighted Zealand’s capability to compete in the obesity market, including its differentiated pipeline, its partnerships with Roche and Boehringer Ingelheim (BI), and its robust cash position of $2.5 billion.
- Then, global pharmaceutical giant AbbVie (market cap $390 billion) gave more detail on its expressed interest in entering the obesity arena. In March 2025, AbbVie entered a partnership with Gubra to develop and commercialize a long-acting amylin analog, GUB014295, for obesity. Mr. Michael said that amylin agonist class is promising due to its favorable tolerability profile. AbbVie will continue to expand its obesity portfolio beyond GUB014295 through business development and licensing strategy and plans to differentiate its portfolio with improved tolerability and durability of weight loss.
- In tech, financial and pipeline updates abounded.
- Omada Health announced preliminary, unaudited 4Q25 results of $257 million in 2025 revenue, up over 50% from 2024. Preliminary 4Q25 revenue is expected to be $72-$74 million, up 50-54% from 4Q24 and 6-9% sequentially. Omada also reported a very healthy adjusted gross margin of just over 66% and CEO Mr. Sean Duffy reiterated the company’s commitment to reaching 70% gross margin. Notably, Omada shared that nearly half of new deals closed were multi-program rather than single-program sales, a positive trend the company continues to see in its most recent deals.
- Mr. Dev Kurdikar, CEO of embecta, expressed confidence in the long-term stability of the needle and pen needle market and showcased the company’s durable, recurring revenue base despite potential shifts driven by increased adoption of insulin pumps and GLP-1 RAs in the US. It was interesting to hear that pen needle revenue has remained stable (approximately +0.6% CAGR) while safety pen needles have grown (+4.9% CAGR). Syringe needle revenue, which represents a smaller portion of the portfolio (~15%) has declined (-4.5% CAGR). On the therapy front, Mr. Kurdikar reiterated that GLP-1 RAs could represent a more than $100 million annual revenue opportunity for embecta by 2033. We look forward to seeing more growth in this important arena.
- Today’s lunchtime keynote featured United States Commissioner of Food and Drugs Dr. Marty Makary, who gave a fascinating talk on multiple initiatives at the FDA. He highlighted proton beam radiotherapy, an advanced cancer treatment, gene therapies, and robotic surgery as just a few examples of the US’s incredible innovation in medicine. However, he said that the system often does incredible work for no credit. When evaluating the success of the system not by its innovation but rather by population health outcomes, Dr. Makary posited that the system is at a fifty-year low. Rates of chronic diseases have sharply increased, and new treatments for diseases have not emerged fast enough to keep pace. “Why does it take 12 years for the FDA to bring drugs from invention to market?” he asked. His compelling talk laid out why 12 years is the current state of affairs and his efforts to change this statistic at the FDA.
Table of Contents
-
Diabetes Therapy
- 1. Zealand Pharma: Dr. Adam Steensberg highlights survodutide and petrelintide; clinical data expected in 2026
- 2. Amphastar: Dr. Jack Zhang shares updates on glucagon, GLP‑1 RAs, and insulin biosimilars
- 3. Sana Biotechnology: Continuing strides for allogeneic islet cell therapy with developed with hypoimmune technology in people with T1D
- 4. Biomea Fusion: Phase 2 data for Icovamenib and phase 1 results of GLP-1 RA BMF-650 expected in 2026
- 5. AbbVie: Interest in entering obesity arena by leveraging familiarity with cash-pay channel and compounding market
- Diabetes Technology
-
Diabetes Big Picture
- 8. The FDA’s Dr. Marty Makary on optimized regulation, accelerating the 12-year approval timeline, and additives in our food supply
- 9. From agentic AI to longevity: ARPA-H’s strategy for breakthrough health innovation
- 10. Employer coverage at crossroads: Managing GLP-1 RAs and breakthrough therapies
Diabetes Therapy
1. Zealand Pharma: Dr. Adam Steensberg highlights survodutide and petrelintide; clinical data expected in 2026
In this afternoon session, Zealand CEO Dr. Adam Steensberg presented insights on the obesity market and updates on Zealand’s pipeline of survodutide (dual glucagon/GLP-1 RA) and petrelintide (long-acting amylin analog). He first highlighted Zealand’s capability to compete in the obesity market, including its differentiated pipeline, partnerships with Roche and Boehringer Ingelheim (BI), and robust cash position of $2.5 billion. Moreover, through internal R&D and external partnerships, Zealand plans to bring over 10 clinical programs by 2030. See below for the milestones for 2026:

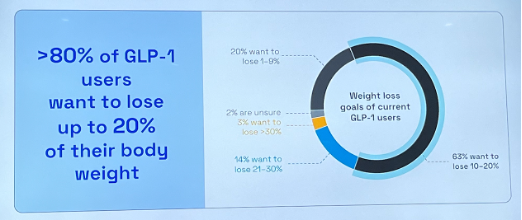
- Dr. Steensberg then delved into the current state of obesity treatments. First, he said that market penetration and maintenance can be expanded, given that 12% of eligible people in the US have been exposed and only ~3% are treated with GLP-1 RAs. Moreover, he pointed to the discrepancy in weight loss efficacy between real world and clinical trials. Although Wegovy and Zepbound conferred 15% and 21% weight loss in STEP-1 and SURMOUNT-1 trials, patients in real-world settings achieved 7.7% and 12.4% weight loss on average, respectively. Dr. Steensberg attributed this difference to: (i) gastrointestinal adverse events intrinsic to GLP-1 RA class; and (ii) different weight loss goals for patients. He emphasized that the field should step back from the “Weight loss Olympics” and understand what the actual medical needs of patients are. According to a survey (n=819) from LifeSci Capital, only about 16% of responders wanted to lose over 20% of their weight. Ultimately, noting that hypertension, dyslipidemia, and T2D each have over eight drug classes, he said that more drug classes are needed for obesity management.
- On petrelintide, he reiterated that the candidate has the best-in-class potential. In a phase 1b trial (n=48), petrelintide conferred weight loss of up to 8.6% (vs. 1.7% with placebo), with only one adverse event-related treatment discontinuation. Results of the ZUPREME-1 trial (n=494) for people with overweight or obesity are expected in 1Q26. Dr. Steensberg said that the ZUPREME-1 trial has a balanced gender distribution (53% vs. 21% female) and higher BMI at baseline (37 vs. 30 kg/m2) compared to the phase 1b trial and is hopeful that the results will be more reflective of the drug’s profile.
- In phase 3 development, Zealand plans to initiate the therapy’s phase 3a program in 2H26. The company will also conduct phase 3b trials to better characterize its effects on cardiovascular (CV) health and other comorbidities. Dr. Steensberg said petrelintide has the potential to confer weight loss of 15-20% and CV risk reduction with less frequent GI events.
- On Zealand’s partnership with Roche, Dr. Steensberg characterized it as an equal collaboration, including 50/50 profit sharing in the US and Europe.
- On survodutide, Dr. Steensberg said that regulatory submissions are expected in 2026 based on the phase 3 SYNCRONIZEprogram – this was great to hear. While he believes the drug may be beneficial in multiple cardiometabolic indications, survodutide has especially demonstrated a best-in-class potential in MASH. In a phase 2 trial, 65% of participants with MASH (vs. 26% on placebo) experienced an improvement in liver fibrosis without the worsening of MASH at Week 48. The phase 3 LIVERAGE program is advancing at full speed for people with MASH and F2-F3 fibrosis or cirrhosis.
- During Q&A, Dr. Steensberg shared that while oral GLP-1 RAs will expand the market and improve manufacturing, they do not resolve the fundamental challenge of GLP-1 RAs: their tolerability profile. He expects that the market will continue to have residual disappointment until novel agents with improved patient experience enter the market. When asked about potential GLP-1 RA and amylin agonist combination therapy that could achieve weight loss >20%, Dr. Steensberg maintained his position that the greater unmet need is the lack of alternatives rather than higher weight loss efficacy. He believes that amylin monotherapy could be used for a broader population who seek improved adherence and maintenance therapy, while combination therapy could target a smaller segment of patients with morbid obesity and/or advanced T2D who are looking for 25-30% weight loss.
2. Amphastar: Dr. Jack Zhang shares updates on glucagon, GLP‑1 RAs, and insulin biosimilars
CEO Dr. Jack Zhang and CFO Mr. Bill Peters highlighted Amphastar’s dual‑strategy growth model, pairing strategic acquisitions like Baqsimi with internally developed complex generics, biosimilars, and novel peptides (see the webcast and presentation slides). The presentation noted that by 2026, proprietary and biosimilar programs will represent 85% of the pipeline, up from 37% in 2021. This shift is supported by sustained R&D investment and expanding technical capabilities across peptides, intranasal delivery, and recombinant DNA biologics.
- Amphastar’s branded pipeline continues to be led by Baqsimi’s (nasal glucagon) projected $250-275 million peak sales and strong US growth following its June 2023 acquisition from Lilly. Mr. Peters noted that only 12% of insulin users currently fill a glucagon prescription, up from 10% at acquisition, leaving substantial room for greater penetration. Internationally, Mr. Peters stated that Amphastar will withdraw from several unprofitable markets after its three-year contractual commitment ends in June 2026, improving the overall margin profile.
- On pipeline updates, AMP‑018, the company’s generic injectable GLP‑1 RA, is planned for a launch in 2027. He said that Amphastar is beginning third‑party sales of GLP‑1 Active Pharmaceutical Ingredient (APIs) in China, marking a strategic shift from internal‑only API use.
- On insulin, Amphastar’s biosimilar insulin programs – AMP‑004 (Novolog) and AMP‑005 (recombinant human insulin) – continue to advance. For AMP‑004, the FDA accepted the company’s biosimilar application in April 2025, and it is currently under review. Mr. Peters noted that while AMP‑025 (insulin degludec) remains in development, Amphastar may pause the program given current market conditions.
3. Sana Biotechnology: Continuing strides for allogeneic islet cell therapy with developed with hypoimmune technology in people with T1D
Dr. Steve Harr (Sana Biotechnology President and CEO) took the stage this morning to present Sana Biotechnology’s vision and progress in the T1D space. Reflecting on 2025, Dr. Harr said the company has made significant strides, including developments across clinical and regulatory conversations for a T1D cure. He emphasized the significant unmet need in people with T1D, who face an immense burden and experience high risks of complications from diabetes. He said, “We can do better.” While islet cell transplantation has been positioned as a promising advancement in the T1D landscape, the procedure requires immunosuppression. To address such challenges, Sana Biotechnology continues advancing UP421, an allogeneic islet cell therapy engineered with the company’s hypoimmune technology. Dr. Harr emphasized that one of the primary distinguishing features of this approach is the elimination of the need for immunosuppression. Throughout the presentation and Q&A, Dr. Harr expressed strong anticipation for the upcoming IND submission for phase 1 trials later this year, as well as insights into the study design.
- Challenges of T1D cure. Dr. Harr said that among the many barriers to developing T1D cures, some of the key challenges include: (i) addressing allogeneic immune rejection without immunosuppression; (ii) developing a “master cell bank” to develop functional beta cells; (iii) manufacturing scale and consistency; and (iv) ensuring purity and potency. Sana Biotechnology has developed UP421 as a single solution that addresses these challenges, and therefore, Dr. Harr affirmed its potential to help the millions of people living with T1D.
- Positive six-month results from the first-in-human study of UP421 transplantation showed efficacy. Building on positive preclinical results, in June 2025, six-month results in one patient showed that UP421 transplantation exhibited a favorable safety profile. Additionally, C-peptide levels and a mixed meal tolerance test showed function and persistence of pancreatic islets. Dr. Harr stated that the patient continues to exhibit detectable C-peptide levels, indicating cell function, one year post-transplantation.
- Dr. Harr affirms Sana Biotechnology’s goal of developing a scalable therapy. While six-month results from the first-in-human study of UP421 represent promising data, Dr. Harr said the company aims to pursue a bigger goal: to develop a scalable treatment. By engineering a universal donor from a single stem cell, the company will apply gene modifications and deliver the therapy as a single treatment.
- Sana Biotechnology’s engagement in several conversations with regulatory bodies. Dr. Harr shared that the company has been in dialogue with multiple regulators worldwide. The FDA has remained “very engaged,” given its understanding of the significant unmet needs in T1D. The regulators have also acknowledged the transformative potential of UP421, especially based on the first-in-human study, and understand the complexity across immunology and stem biology. Across multiple conversations, Dr. Harr said the FDA has been supporting the company in navigating with clear expectations.
- Insights into the phase 1 trial of UP421 transplantation. Dr. Harr said that the phase 1 trial plans to encompass a broad population, starting with adults and later expanding to those at higher risk of comorbidities (e.g., CVD) and younger populations. The trial plans to be conducted in the US and other geographies. Dr. Harr expressed confidence that the phase 1 trial will be straightforward, showing efficacy within just a few weeks of the study.
4. Biomea Fusion: Phase 2 data for Icovamenib and phase 1 results of GLP-1 RA BMF-650 expected in 2026
In this afternoon session, Dr. Mick Hitchcock, interim CEO of Biomea Fusion, highlighted key programs: icovamenib and the GLP-1 RA BMF-650. He first began by emphasizing that current treatments for T2D primarily address downstream metabolic symptoms, rather than the disease pathway. Icovamenib is an oral covalent menin inhibitor that promotes beta cell proliferation and function in people with T2D. This candidate was developed based on human physiology: pregnant or lactating women were found to suppress menin, leading to beta cell expansion and insulin secretion. Preclinical studies in rats and human islet cells confirmed that menin inhibition promotes beta cell proliferation.
- In the phase 2b COVALENT-111 trial, icovamenib significantly reduced A1c values by 1.2 percentage points (vs. a 0.3 percentage point increase with placebo) and increased C-peptide level by 29% (vs. 2%) in the severe insulin-deficient subgroup (~60% of T2D) at Week 52. Dr. Hitchcock pointed out that a short 12-week dosing period induced epigenetic changes for beta cell regeneration, resulting in sustained improvement in A1c values for a year. Moreover, a post-hoc analysis found that icovamenib is especially efficacious in people on GLP-1 RA therapy whose baseline A1c was >7.0%. Icovamenib conferred a 1.2 percentage point reduction in A1c in this population compared to a 0.6 percentage point increase with placebo. This finding showed that Icovamenib was generally well-tolerated, with no reports of adverse event-related discontinuations or serious adverse events. Ultimately, Dr. Hitchcock said that icovamenib could offer patients a short-term oral treatment, rather than lifelong insulin use.
- Looking ahead, Biomea Fusion will evaluate icovamenib in the phase 2b COVALENT-211 trial for people with insulin-deficient T2D (~20% of T2D) and the phase 2 COVALENT 212 trial for people with difficult-to-manage T2D already on GLP-1 RAs (70% of people taking GLP-1 RAs). Both trials will begin recruiting in 1Q26, and 26-week results are expected in 4Q26.
- During Q&A, Dr. Hitchcock said that the “win scenario” for icovamenib would be conferring a ≥0.5% A1c reduction at 26 weeks to remain “approvable.” An A1c reduction by 1.8-2.0% would allow icovamenib to be competitive. He also clarified that the phase 3 trials for icovamenib could be smaller programs, given that the drug is administered for 12 weeks and thus is not considered a “chronic agent.”
- BMF-650 is an oral small molecule GLP-1 RA. It is currently evaluated in the phase 1 GLP-131 trial in people with obesity (BMI ≥30 kg/m2) who are otherwise healthy; 28-day weight loss results are expected in 2Q26. Dr. Hitchcock said that BMF-650 is designed for better bioavailability and has the potential to deliver more consistent efficacy than other oral GLP-1 RAs like orforglipron. Moreover, BMF-650 has the same chemotype as orforglipron, as opposed to the chemotype to which the now-discontinued danuglipron, lotiglipron, and TERN-601 belonged. This chemotype, as well as preclinical studies in monkeys, gives Biomea Fusion confidence that BMF-650 will not cause liver-related safety issues.
- During Q&A, Dr. Hitchcock defined success for BMF-650 as involving a competitive weight loss magnitude, favorable tolerability, a faster titration scheme, and durability of weight loss.
5. AbbVie: Interest in entering obesity arena by leveraging familiarity with cash-pay channel and compounding market
In this morning session, CEO Mr. Robert Michael shared updates on AbbVie. As background, AbbVie is a global pharmaceutical company with a market cap of $390 billion. Its focus areas span immunology, oncology, neuroscience, ophthalmology, aesthetics, and infectious diseases. Blockbuster drugs include autoimmune disease medications Humira (adalimumab) –which is now facing loss-of-exclusivity – IL-23 inhibitor Skyrizi (risankizumab-rzaa), and JAK inhibitor Rinvoq (upadacitinib). Botox (onabotuliumtoxinA) used for cosmetic purposes is also a multi-billion-dollar product made by AbbVie.
- AbbVie expresses interest in entering the obesity arena. In March 2025, AbbVie entered a partnership with Gubra to develop and commercialize a long-acting amylin analog, GUB014295, for obesity. Mr. Michael said that amylin agonist class is promising due to its favorable tolerability profile. AbbVie will continue to expand its obesity portfolio beyond GUB014295 through business development and licensing strategy and plans to differentiate its portfolio with improved tolerability and durability of weight loss. Moreover, Mr. Michael believes that AbbVie is well-positioned especially given its familiarity with the cash-pay channel and compounding market. Notably, the compounding business had become the second largest segment in the aesthetics market “over a matter of a few quarters.”
- As a reminder, the candidate conferred dose-dependent weight loss up to 3% at six weeks, compared to 1% weight gain with placebo, in a phase 1 SAD trial. The candidate is currently evaluated in a phase 1 multiple ascending dose (MAD) study (n=124), which is recruiting participants and is estimated to complete in April 2026. While early, Mr. Michael said that the candidate has a potential to be “differentiated amylin,” and is eager to see more data.
Diabetes Technology
6. Omada Health: Preliminary full-year 2025 revenue of ~$257 million (+52%); 2026 will see investment in GLP-1 RA Care Track and AI tools
Omada Health announced preliminary, unaudited 4Q25 results on the first day of JPM 2026. The company reported approximately $257 million in 2025 revenue, up 52% from 2024. Preliminary 4Q25 revenue is expected to be $72-$74 million, up 50-54% from 4Q24 and 6-9% sequentially. Omada also reported an adjusted gross margin of 66.4% and today, CEO Mr. Sean Duffy reiterated the company’s commitment to reaching 70%. Notably, Omada shared that nearly half of new deals closed were multi-program rather than single-program sales, a trend the company continues to see in its most recent deals. Omada now serves approximately 2,000 customers across employers, health plans, and integrated health systems, covering 886,000 members, up 55% from 2024. Beyond what Mr. Duffy described as the “two G’s of great growth” reflected in Omada’s 4Q25 performance, he also discussed the company’s focus on an additional “two G’s”: GLP-1s and GPTs.
- Mr. Duffy reiterated Omada’s mission to “bend the curve” of chronic disease. The company began with prevention and weight health and has since expanded into diabetes, hypertension, and musculoskeletal care, reflecting the high prevalence of comorbidities (e.g., 74% of people with diabetes also have hypertension). Omada’s programs integrate connected devices – including cell-connected weight scales, blood pressure monitors, and CGMs – with in-app goal setting, clinical visits, community engagement, and progress tracking. More than 20 million people now have benefits coverage for at least one Omada program. As of December 2024, 55% of members maintained at least monthly engagement one year into the program, declining modestly to 50% at two years. Omada has published more than 30 peer-reviewed studies demonstrating clinical and economic outcomes and continues to focus on securing accreditations, including National Committee for Quality Assurance (NCQA) accreditation for diabetes and hypertension population health.
- Mr. Duffy spoke extensively about Omada’s GLP-1 RA Care Track, describing GLP-1 RAs as “a moment to seize.” He positioned the offering as a “GLP-1 RA maximizer,” designed to enhance weight loss during therapy and minimize weight regain after discontinuation. For example, Omada reported internal data showing 28% greater weight loss among participants using its Enhanced GLP-1 RA Care Track and just 0.8% weight regain one year after stopping therapy, compared with 11-12% weight regain observed in studies such as SURMOUNT-4 (Zepbound) and STEP-1 (Wegovy). He noted that Omada recently began prescribing GLP-1 RAs and expects an increasingly complex landscape with the introduction of oral therapies and additional single- and dual-agonist therapies.
- Addressing the potential impact of oral GLP-1 RAs, Mr. Duffy said that more therapeutic options typically drive greater utilization and emphasized Omada’s ability to help identify which patient profiles are best suited for oral versus injectable therapies. Management also pushed back on the notion that lower-priced oral GLP-1 RAs would diminish Omada’s relevance, emphasizing that employer goals center on the additive benefits of Omada’s tools. Omada Health President Mr. Wei-Li Shao also explained that their entrance into prescribing GLP-1 RAs was driven by employers and health plan customers wishing their members did not experience a gap between therapy initiation and Omada’s digital support system. When asked about future prescribing, he indicated Omada would consider additional medications for other chronic conditions over time.
- On the technology front, Mr. Duffy highlighted its growing use of AI under the OmadaSpark umbrella, including a nutritional intelligence agent that is increasingly being used by members today.
- Looking ahead, Omada plans to expand its addressable market in covered lives, improve enrollment by converting eligible individuals into active members, and drive stronger retention and engagement, supported by continued product innovation.
7. embecta: GLP-1 RAs offer >$100 million annual opportunity; “market-appropriate” products for China and other geographies
Mr. Dev Kurdikar, CEO of embecta, expressed confidence in the long-term stability of the needle and pen needle market and showcased the company’s durable, recurring revenue base despite potential shifts driven by increased adoption of insulin pumps and GLP-1 RAs in the US. Pen needle revenue has remained stable (approximately +0.6% CAGR) and safety pen needles have grown (+4.9% CAGR). While syringe needle revenue has declined (-4.5% CAGR), it represents a smaller portion of the portfolio (~15%).
- As he shared on prior earnings calls, Mr. Kurdikar reiterated that GLP-1 RAs could represent a more than $100 million annual revenue opportunity for embecta by 2033. Much of this opportunity stems from generic GLP-1 RAs, which are typically delivered in pens co-packaged with pen needles. Generic manufacturers are seeking to replicate the packaging of branded products such as Ozempic, creating a natural B2B opportunity for embecta. More than one-third of embecta’s potential generic partners (over 30 companies) are either in contract negotiations or have executed agreements with it already, with some already having placed purchase orders. These partners anticipate launches in Canada, Brazil, China, and India beginning in 2026, with developed markets in Europe and the US coming as soon as 2031-2032. While branded GLP-1 RAs are not currently co-packaged with pen needles, embecta has developed combination packs and is in active discussions with branded pharmaceutical companies. embecta does not see a role in GLP-1 RA autoinjectors.

- Mr. Kurdikar also addressed embecta’s operations in China, which (together with Hong Kong and Taiwan) accounts for ~10% of total revenue. embecta distributes through three national distributors in China, but shifting geopolitical dynamics in 2025 drove increased preference for local Chinese brands, along with heightened price competition. In response, embecta introduced products at more competitive price points to better match local market dynamics. While stabilization is expected to take several quarters, management remains confident in China’s long-term importance.
- He also discussed embecta’s strategy to develop additional “market-appropriate” pen needles and syringes for other price-sensitive regions. The company previously exited certain tender-driven markets, such as parts of Latin America, rather than significantly discount its “premium products.” It is now preparing to reenter these markets with cost-optimized offerings better aligned with local needs. embecta has completed design for these products, installed their manufacturing equipment, and is currently validating production lines, with launches expected in the near to medium term in markets where its current share is <5%. These products are intended to drive incremental volume, leveraging embecta’s historical familiarity with these markets.
- Looking ahead, Mr. Kurdikar framed 2025-2028 as a period focused on strengthening embecta’s core business, expanding the product portfolio, and building financial flexibility. He cited the discontinuation of the company’s patch pump program last year due to capital allocation priorities and the elimination of nearly $185 million in debt during 2025 as representative of this last point. He also highlighted the completion of embecta’s brand transition in the US and Canada, featuring refreshed packaging and the replacement of the “BD” name with “embecta” to deepen engagement with customers and healthcare providers. This rebranding effort is underway globally and is expected to be completed in most markets by calendar year 2026, with a small number of Asian markets to follow. From 2028 onward, embecta aims to evolve into “a broad-based medical supplies company” serving both chronic care patients and drug delivery partners.
Diabetes Big Picture
8. The FDA’s Dr. Marty Makary on optimized regulation, accelerating the 12-year approval timeline, and additives in our food supply
United States Commissioner of Food and Drugs Dr. Marty Makary gave a strong talk discussing the FDA’s balance between regulation and stimulating innovation. He spoke of the privilege that it is to lead the FDA and to address attendees at the conference today, saying that the US healthcare system is amazing. He highlighted proton beam radiotherapy, an advanced cancer treatment, gene therapies, and robotic surgery as just a few examples of the US’s incredible innovation in medicine. However, he said that the system often does incredible work for no credit. When evaluating the success of the system not by its innovation but rather by population health outcomes, Dr. Makary said that the system is at a fifty-year low. Rates of chronic diseases have sharply increased, and new treatments for diseases have not emerged fast enough to keep pace. “Why does it take 12 years for the FDA to bring drugs from invention to market?” he asked. His compelling talk laid out why 12 years is the current state of affairs and his efforts to change this statistic at the FDA.
- In 2026, the FDA will remove onerous requirements that do not result in safer medications, seeking a process that scrutinizes safety, when necessary, yet makes more medications available without prescriptions. He described the roadblocks to availability that many therapeutics face both before and after approval, with extensive trial requirements that do not result in more robust data and prescription requirements for therapies without strong risk factors. Dr. Makary said that the FDA will implement the use of Bayesian statistics to increase the statistical power of studies and streamline them, with one pivotal trial becoming the new standard, as opposed to two. The FDA will also work to eliminate unnecessary animal testing, which comes with animal cruelty concerns and can cause treatment delays. As it stands, each monoclonal antibody that is approved requires 144 chimpanzees to be tested – at a very high logistical and ethical cost.
- In June 2025, the FDA launched the Commissioner's National Priority Voucher (CNPV) Pilot Program, which will reduce drug and biological product application or efficacy supplement approval times for key therapies to 1-2 months from the current 10-12 months. In conjunction with Most-Favored-Nation pricing established by the White House in May 2025 to lower drug costs for Americans, the FDA seeks true therapeutic accessibility in both development and sales. “A 100% effective therapy that only 50% of people can afford is only 50% effective,” Dr. Makary said.
- Dr. Makary said that no patients should visit emergency rooms for prescription refills due to administrative lag and hopes to improve accessibility in all ways. Many patients find themselves unable to schedule timely appointments to receive prescription refills, forcing them to visit emergency rooms seeking medication, which adds further strain to the health system. Safe medications with no abuse potential and no need for continued laboratory work to monitor response should be available over the counter, said Dr. Makary, adding that the FDA must move away from a paternalistic mindset.
- The FDA plans to review over 1,000 chemicals found in the US food supply that have been banned in other nations. In April 2025, the FDA announced that petroleum-based synthetic dyes including Red No. 40 will be phased out of US foods by the end of 2026. Dr. Makary said that this decision had been in discussion for nearly 35 years yet had been thwarted by administrative hurdles. As part of the Make America Healthy Again (MAHA) movement, the FDA will review chemical additives to the US food supply and plans to eliminate the Generally Recognized As Safe (GRAS) loophole in 2026. The policy allows food manufacturers to declare new additives as safe without FDA review, which may lead to unknowingly harmful chemicals in the food supply.
- To achieve these initiatives, the FDA has begun hiring over 1,000 new scientists. Dr. Makary said that 450 of these scientists are currently being onboarded, and that recent layoffs at the FDA did not affect scientists and were focused on streamlining administrative roles. However, Skadden, Arps, Slate, Meagher & Flom LLP and Affiliates previously reported that scientists involved in the regulatory process were affected by these layoffs.
- “The trains are running on time, but the process needs to be reformed,” said Dr. Makary. He seeks deep, systemic change to the FDA’s processes to modernize and support the scientific community. Only then will we be able to deliver more treatments and healthier food for America’s children.
9. From agentic AI to longevity: ARPA-H’s strategy for breakthrough health innovation
Dr. Alicia Jackson, Director of ARPA-H (the Advanced Research Projects Agency for Health), highlighted several of the agency’s accomplishments in 2025 and outlined priorities for the years ahead. As background, ARPA-H is a federal health innovation agency with approximately $1.5 billion in annual funding dedicated to high-impact advances in human health. Unlike traditional grant-making organizations, ARPA-H operates through milestone-driven contracts. Dr. Jackson said that ARPA-H also acts as a “benevolent dictator,” allowing dissonance within the organization in how to approach a given project rather than seeking consensus, which she explained as an intentional strategy to accelerate breakthrough innovation. The agency works closely with the FDA to help navigate regulatory risk, creating pathways that others in the ecosystem can follow. Namely, Dr. Jackson said that ARPA-H is willing to take early, high-risk bets in partnership with the FDA to enable later entrants to scale and succeed.
- Dr. Jackson highlighted yesterday’s launch of ADVOCATE (Agentic AI-EnableD CardioVascular CAre TransfOrmation), an agentic AI initiative focused on cardiovascular disease management. Broadly, she described AI as foundational to nearly every ARPA-H program, noting the agency’s unique ability to integrate large, fragmented datasets across the healthcare system. ADVOCATE aims to support both biotech innovation and clinical care, particularly in regions with primary care and specialist shortages. By embedding medical intelligence into AI-powered care delivery and leveraging wearables to monitor patients remotely, these tools could help determine when patients can safely recover at home versus requiring in-person care at a medical center. ARPA-H project manager Dr. Haider Warraich commented that this technology is not yet ready for widespread deployment, but ARPA-H is working to define regulatory pathways, post-approval monitoring frameworks, and strategies to scale it in order to support future adoption.
- Dr. Jackson also highlighted the Biological Data Fabric, an initiative designed to unify fragmented medical data, including EHRs and pathology reports, across health systems to create longitudinal patient histories. Not only does the technology allow families and clinicians to access comprehensive patient histories, but it also allows them to compare their medical journeys to others and learn from comparable cases to inform treatment decisions. The approach has already been successfully implemented in two hospital systems and is now being expanded to across 200 pediatric brain cancer centers – treating the leading cause of disease-related death in children – with $50 million in funding from ARPA-H.
- Speaking on another AI-enabled effort, the CATALYST program, ARPA-H program manager Dr. Andrew Kilianski explained the mission to reduce reliance on animal models by using in silico approaches in drug development. These models have the potential to significantly accelerate clinical trial timelines and reduce development costs.
- Dr. Jackson also discussed ARPA-H’s growing focus on longevity and age reversal. Aging is the greatest risk factor for most chronic and degenerative diseases, and even modest reductions in the biological age of key organ systems like the brain or heart could generate trillions of dollars in system savings. ARPA-H is investing in early-stage research to identify causal biomarkers of aging and link them directly to disease, helping the broader field prioritize the most promising therapeutic pathways. This includes leveraging resources such as the Interventions Testing Program and mouse model data to evaluate potential longevity therapies.
- Finally, Dr. Jackson addressed ARPA-H’s genetic medicines portfolio, citing the high-profile “Baby KJ” case as proof of the technology’s potential. While the science is promising, current approaches are not scalable to all patients who could benefit. Therefore, ARPA-H is focused on reducing manufacturing, delivery, and storage costs through innovations in extracellular vesicles, improved AAV vectors, continuous manufacturing, and better storage options.
10. Employer coverage at crossroads: Managing GLP-1 RAs and breakthrough therapies
Mr. Brian Heath (Kite Pharma), Dr. Will Shrank (Aradigm), and Dr. Dan Knecht (EmblemHealth) discussed strategies to reduce the cost of employer-sponsored coverage for high-cost therapies. The panel discussed how rising spending on GLP-1 RAs and the complexity of cell and gene therapies can strain this coverage and highlighted gaps in population-level management and payment certainty. They agreed that expanding access will require greater alignment across payers, providers, and manufacturers.
- The panel addressed the GLP-1 RA class that now accounts for ~15% of total spending among employers that offer open access. Dr. Shrank emphasized that GLP-1 RAs are transformative therapies should be available to all patients who need them, but acknowledged that universal access under current pricing would be prohibitively expensive. He commented on unexpected market dynamics, including direct-to-consumer models and compounded versions of GLP-1s RA, that he did not anticipate would gain such traction. Overall, he characterized the space as a “natural experiment” featuring widespread innovation and ongoing price compression as negotiations evolve.
- Dr. Knecht described the healthcare ecosystem as having been “on our heels rather than on our toes” when the latest generation of GLP-1 RAs launched. In hindsight, he said the ecosystem could have moved more quickly to anticipate demand and to counter longstanding narratives that frame obesity as a moral failing rather than a medical condition. Such efforts could have enabled broader coverage and more proactive adoption – though he also acknowledged early limitations with supply constraints.
- Dr. Shrank added that high-cost drugs covered under the medical benefit represent an under-managed category in healthcare, despite them being some of the most clinically impactful medications. He said that carriers have limited tools today but argued that a more proactive approach that “matches the right drug to the right patient” represents a clear opportunity for disruption.
- The panel also spent considerable time discussing access to cell and gene therapies. Mr. Heath explained that challenges in this space extend well beyond cost. Providers face uncertainty around the amount and timing of reimbursement – payment can be negotiated or delayed for months after the fact. Dr. Shrank agreed, noting that this dynamic has made it difficult for manufacturers to recoup R&D investment. Dr. Knecht warned that if these issues persist, more small and mid-sized employers may opt out of covering these therapies altogether.
- Despite these challenges, Mr. Heath emphasized that adoption is meaningful: more than 30,000 patients globally have received cell therapies. Kite Pharma operates in over 40 countries with more than 570 authorized treatment centers and participates in the CAR-T Vision initiative, which brings together stakeholders across the healthcare ecosystem. Mr. Heath identified three barriers hindering access: (i) lack of awareness among patients and providers; (ii) limited access due to insufficient accredited treatment centers; and (iii) the need for improved financing models with faster coverage and reimbursement decisions.
- On the policy front, Dr. Shrank highlighted CMS’s growing engagement in cell and gene therapy reimbursement as an encouraging sign. He pointed to CMS’s value-based contracting approach for sickle cell disease, which aligns incentives among manufacturers, payers, and patients by defining outcomes collaboratively and allowing states to opt in – an option already adopted by roughly two-thirds of states. He suggested this framework could extend to other conditions, noting that approximately 40 cell and gene therapies are currently approved and currently an estimated 3,000-4,000 in development, including drugs for more prevalent diseases like T1D. While an important step forward, he emphasized that this approach alone is not a comprehensive solution.
The panel also spoke more broadly on improving access to expensive therapies that target smaller patient populations. They came to consensus around the desire to shift away from fee-for-service payment models and toward more outcomes-based arrangements. Dr. Shrank noted that for potentially curative gene therapies, outcomes are often easier to define. Dr. Knecht added that these agreements can be especially impactful when evidence is still emerging, as they help catalyze initial access. However, as the body of evidence grows and the value of the therapies becomes clearer, more traditional fee-for-service pricing models often become more practical.
-- by Kayla Mathieu, Esther Min, Jeremy Alkire, Nour Khachemoune, Kat Moon, Monica Oxenreiter, and Kelly Close